Difference between revisions of "AGiR! for genomic integrity/cometassaydev"
| (39 intermediate revisions by 3 users not shown) | |||
| Line 1: | Line 1: | ||
| − | This | + | This page describes some concrete aspects of the cheek cell comet assay protocol. <br> |
| − | [[File:CheekCellComet 2dec16slide2x10GFPb.jpg]] | + | [[File:CheekCellComet 2dec16slide2x10GFPb.jpg|thumb|upright=0.9|Illustration of a "comet", the migration of the DNA after an electrophoresis]] |
==Experiments== | ==Experiments== | ||
* Cheek Cell comets | * Cheek Cell comets | ||
| + | * Test of [[Tumeric | Tumeric for staining]] on cheek cells (7avr17) | ||
| + | * Test of [[SDSorDishSoapforPK | SDS vs Dish soap vs Standard]] protocol | ||
==Conditions for Comet Cell Assays == | ==Conditions for Comet Cell Assays == | ||
| Line 11: | Line 13: | ||
*tests of pbs vs saline | *tests of pbs vs saline | ||
*tests for optimal numbers of cells | *tests for optimal numbers of cells | ||
| − | *tests of filtration vs gravity for eliminating big clumps of cells (1st attempt with lab 40micron filter run 8march2017, with Anna and | + | *tests of filtration vs gravity for eliminating big clumps of cells (1st attempt with lab 40micron filter run 8march2017, with Anna and Bastien) |
*tests for staining dyes | *tests for staining dyes | ||
*control conditions - negative control with no protease treatment, positive control with H2O2 | *control conditions - negative control with no protease treatment, positive control with H2O2 | ||
*tests of non-lab 'kitchen sink' components for reactions (dish soap, contact lens cleaner, etc...) | *tests of non-lab 'kitchen sink' components for reactions (dish soap, contact lens cleaner, etc...) | ||
| + | *tests of lab water vs bidistilled water | ||
==Current 'best' protocol== | ==Current 'best' protocol== | ||
| Line 25: | Line 28: | ||
3) embed cells in 1% low melting point agarose (made up in standard saline solution) | 3) embed cells in 1% low melting point agarose (made up in standard saline solution) | ||
| − | aiming for about 20,000 cells to be added to 100 microliters agarose (kept at 42<sup>o</sup>C)<br> | + | aiming for about 20,000 cells to be added to 100 microliters molten agarose (kept at 42<sup>o</sup>C)<br> |
mix and put on clean microscope slide using a clean coverslip to flatten it in place. (can flatten the cell suspension on top of a pre-made agarose base, if desired, see below)<br> | mix and put on clean microscope slide using a clean coverslip to flatten it in place. (can flatten the cell suspension on top of a pre-made agarose base, if desired, see below)<br> | ||
| − | 4) treat cell patches with first solutions: saline only, for test cases (3-5 replicates ideally); and H2O2 (0.01%) for positive control case.<br> | + | 4) treat cell patches with first solutions: saline only, for test cases (3-5 replicates ideally); and H2O2 (0.01%) for positive control case. After 5min pre-treatment, wash twice with saline solution<br> |
| − | 5) equilibrate all cell patches with Proteinase K (PK) buffer (for 2ml: 40 microliters 0.5M Tris pH8, 400 microliters 0.5M EDTA, 1ml 5M NaCl, | + | 5) equilibrate all cell patches with Proteinase K (PK) buffer<br> |
| + | Final Concentrations: <br> | ||
| + | 2.5M NaCl <br> | ||
| + | 0.1M EDTA <br> | ||
| + | 10mM Tris (pH 8) <br> | ||
| + | 1% Triton x100 <br> | ||
| + | |||
| + | (for 2ml: 40 microliters 0.5M Tris pH8, 400 microliters 0.5M EDTA, 1ml 5M NaCl, 200 microliters 10% Triton, 0.36ml H2O) <br> | ||
x2 changes (drops of liquid, about 20-50 microliters depending on patch sizes) <br> | x2 changes (drops of liquid, about 20-50 microliters depending on patch sizes) <br> | ||
| Line 36: | Line 46: | ||
''keep at least one cell patch without this treatment as your negative control!''<br> | ''keep at least one cell patch without this treatment as your negative control!''<br> | ||
RT incubation (watch under scope to see cells 'disappearing' leaving nucleoids - usually only about 20 minutes max)<br> | RT incubation (watch under scope to see cells 'disappearing' leaving nucleoids - usually only about 20 minutes max)<br> | ||
| + | ''Both Detergent and PK are essential for this to occur without induction of apoptosis, which would ruin any hope for successful observation of comets.'' <br> | ||
| + | This works well over night in the refrigerator, also, as confirmed in November 2019 expts. | ||
7) as treatment seems sufficient, apply 1X TBE pH9 (alkaline treatment) to cell patches<br> | 7) as treatment seems sufficient, apply 1X TBE pH9 (alkaline treatment) to cell patches<br> | ||
| Line 45: | Line 57: | ||
9) run the electrophoresis step: 12V 15 min (only 2 slides will fit easily in the gel box currently used in the lab, but other possibilities exist) <br> | 9) run the electrophoresis step: 12V 15 min (only 2 slides will fit easily in the gel box currently used in the lab, but other possibilities exist) <br> | ||
| − | 10) staining and imaging (most success with SYBR safe dye, diluted 1/10,000 in TBE for staining | + | 10) staining and imaging (most success with SYBR safe dye, diluted 1/10,000 in TBE for staining) and epifluor imaging ('green' channel)<br> |
==Ideas for protocol improvement== | ==Ideas for protocol improvement== | ||
| − | + | For running the comet assay, leaving the treatment overnight (PK buffer etc) has given very good results (in particular with Violaine, Vithoo and Jennifer in the summer of 2018). They also showed clearly that the lab water used is critically important. Just using distilled water from the shop has the potential to really increase apoptosis in the agarose patches, leading to 'no comet' results. <br> | |
| + | Additionally, so the patches stay flat during the run, it is a good idea to place the patches between 2 slides, as also successfully demonstrated in the summer of 2018. <br> | ||
| + | Here are pages they put up about their work:<br> | ||
| + | ''For the 2018 progress about genomic integrity, see [[Genomic Integrity 2018]] and for the DIY Comet Cell Assay see [[Comet Cell Assay 2018]].'' | ||
| + | <br< | ||
| + | Ultimately, the 'cheek cell chip' (more on this below) is hoped to make everything easy, to avoid having to do so many manipulations (even with paint-brushes, to get patches of cells mounted on slides for imaging). <br> | ||
| + | While we had tried to assess alternatives that would form a nicely colored DNA 'comet tail' for easier DIY image acquisition/analyses, but neither Hemalum after a MeOH:acetic acid (3:1) fix nor methylene blue aqueous seem very useful at all in this regard... Some tests with methylene blue and iodine have been a bit promising, but further tests were generally not encouraging. New ideas are very welcome!<br> | ||
| + | <br> | ||
| + | In the meantime, DIYmicroscopes that can use a blue LED excitation light are being worked upon, too!! The [https://openflexure.org/ OpenFlexure] system for epifluor may just do the trick, we hope. | ||
| + | A second build to their v6 still needs to be put together with the motors, and then we may really be golden, fingers crossed.<br> | ||
| + | <br> | ||
| + | If you have trouble removing the coverslip from the patch of embedded cells without messing it up, you can add buffer first, or lose patches during manipulations, you can make an initial patch on the slide before adding the cell suspension in agarose. <br> | ||
| + | Use two slides covered with tape and 2 more slides, as in [http://www.wormbook.org/chapters/www_intromethodscellbiology/cellfig1.jpg Monica Driscoll's old WormBook method]. (of course, instead of worms in solution, put your embedded cheek cells in molten agarose on top of pre-made patch, then flatten with coverslip as above. <br> | ||
<br> | <br> | ||
| − | |||
| − | |||
| − | |||
One patch can be cut into sections, for quick tests in different conditions, for example: middle untreated, one side for test, other side for positive control.<br> | One patch can be cut into sections, for quick tests in different conditions, for example: middle untreated, one side for test, other side for positive control.<br> | ||
<br> | <br> | ||
| − | == Ideas for ' | + | == Ideas for 'Cheek Cell Chip' development as an 'automated' system == |
| − | an open source microfluidic device | + | The idea is to make an open-source microfluidic device or something inspired by microfluidics. The chip should be easy to make and to use. All the comet cell treatments should be carried out on the chip, thus contain electrodes, etc... The dream configuration would allow micronuclei assays to be done first, and comets later on the same embedded cells. In the context of the OpenFlexure scope, we may simply modify the upper stage attachment, to become the 'cheek cell chip' in fact. But first we need to see more than just nuclei with this microscope/RaspberryPi system (work in progress!)! |
Latest revision as of 11:27, 16 January 2021
This page describes some concrete aspects of the cheek cell comet assay protocol.

Experiments
- Cheek Cell comets
- Test of Tumeric for staining on cheek cells (7avr17)
- Test of SDS vs Dish soap vs Standard protocol
Conditions for Comet Cell Assays
- tests of pbs vs saline
- tests for optimal numbers of cells
- tests of filtration vs gravity for eliminating big clumps of cells (1st attempt with lab 40micron filter run 8march2017, with Anna and Bastien)
- tests for staining dyes
- control conditions - negative control with no protease treatment, positive control with H2O2
- tests of non-lab 'kitchen sink' components for reactions (dish soap, contact lens cleaner, etc...)
- tests of lab water vs bidistilled water
Current 'best' protocol
1) isolate cells in saline solution (0.9% NaCl in water) after gentle toothbrush collection from inner cheek (rinse mouth with fresh water before 'harvest')
- about 10ml solution
Let biggest clumps settle away, and take remaining solution to pellet most cells
(2ml eppi tube, 2k rpm, 2min), repeating until all solution is pelleted.
Resuspend pellets in about 200 microliters of fresh saline and estimate cell concentration and quality under scope (can use hemocytometer for counting)
3) embed cells in 1% low melting point agarose (made up in standard saline solution)
aiming for about 20,000 cells to be added to 100 microliters molten agarose (kept at 42oC)
mix and put on clean microscope slide using a clean coverslip to flatten it in place. (can flatten the cell suspension on top of a pre-made agarose base, if desired, see below)
4) treat cell patches with first solutions: saline only, for test cases (3-5 replicates ideally); and H2O2 (0.01%) for positive control case. After 5min pre-treatment, wash twice with saline solution
5) equilibrate all cell patches with Proteinase K (PK) buffer
Final Concentrations:
2.5M NaCl
0.1M EDTA
10mM Tris (pH 8)
1% Triton x100
(for 2ml: 40 microliters 0.5M Tris pH8, 400 microliters 0.5M EDTA, 1ml 5M NaCl, 200 microliters 10% Triton, 0.36ml H2O)
x2 changes (drops of liquid, about 20-50 microliters depending on patch sizes)
6) treat cells with PK+ solution (final PK concentration at 0.5mg/ml: 5 microliters of 20mg/ml PK stock in 200 microliters PK buffer)
keep at least one cell patch without this treatment as your negative control!
RT incubation (watch under scope to see cells 'disappearing' leaving nucleoids - usually only about 20 minutes max)
Both Detergent and PK are essential for this to occur without induction of apoptosis, which would ruin any hope for successful observation of comets.
This works well over night in the refrigerator, also, as confirmed in November 2019 expts.
7) as treatment seems sufficient, apply 1X TBE pH9 (alkaline treatment) to cell patches
2x 5 min
8) equilibrate all patches with normal 1X TBE
3x 2 min
9) run the electrophoresis step: 12V 15 min (only 2 slides will fit easily in the gel box currently used in the lab, but other possibilities exist)
10) staining and imaging (most success with SYBR safe dye, diluted 1/10,000 in TBE for staining) and epifluor imaging ('green' channel)
Ideas for protocol improvement
For running the comet assay, leaving the treatment overnight (PK buffer etc) has given very good results (in particular with Violaine, Vithoo and Jennifer in the summer of 2018). They also showed clearly that the lab water used is critically important. Just using distilled water from the shop has the potential to really increase apoptosis in the agarose patches, leading to 'no comet' results.
Additionally, so the patches stay flat during the run, it is a good idea to place the patches between 2 slides, as also successfully demonstrated in the summer of 2018.
Here are pages they put up about their work:
For the 2018 progress about genomic integrity, see Genomic Integrity 2018 and for the DIY Comet Cell Assay see Comet Cell Assay 2018.
<br<
Ultimately, the 'cheek cell chip' (more on this below) is hoped to make everything easy, to avoid having to do so many manipulations (even with paint-brushes, to get patches of cells mounted on slides for imaging).
While we had tried to assess alternatives that would form a nicely colored DNA 'comet tail' for easier DIY image acquisition/analyses, but neither Hemalum after a MeOH:acetic acid (3:1) fix nor methylene blue aqueous seem very useful at all in this regard... Some tests with methylene blue and iodine have been a bit promising, but further tests were generally not encouraging. New ideas are very welcome!
In the meantime, DIYmicroscopes that can use a blue LED excitation light are being worked upon, too!! The OpenFlexure system for epifluor may just do the trick, we hope.
A second build to their v6 still needs to be put together with the motors, and then we may really be golden, fingers crossed.
If you have trouble removing the coverslip from the patch of embedded cells without messing it up, you can add buffer first, or lose patches during manipulations, you can make an initial patch on the slide before adding the cell suspension in agarose.
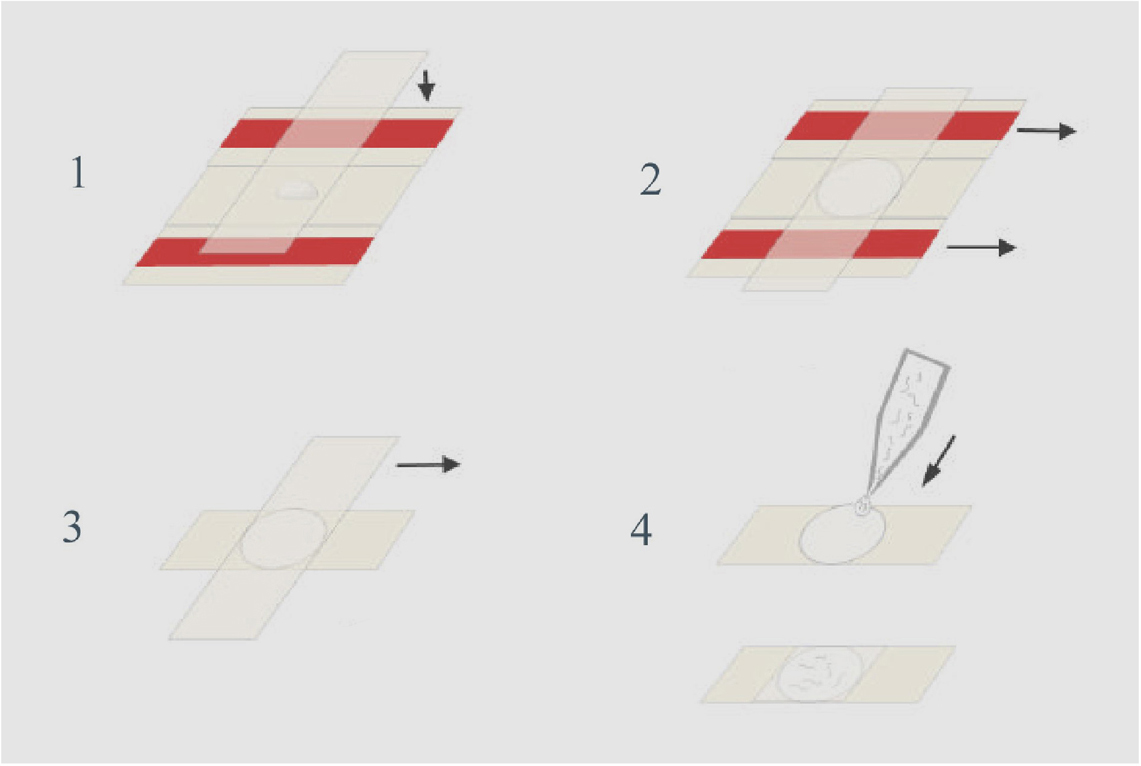
Use two slides covered with tape and 2 more slides, as in Monica Driscoll's old WormBook method. (of course, instead of worms in solution, put your embedded cheek cells in molten agarose on top of pre-made patch, then flatten with coverslip as above.
One patch can be cut into sections, for quick tests in different conditions, for example: middle untreated, one side for test, other side for positive control.
{kind=link}
Ideas for 'Cheek Cell Chip' development as an 'automated' system
The idea is to make an open-source microfluidic device or something inspired by microfluidics. The chip should be easy to make and to use. All the comet cell treatments should be carried out on the chip, thus contain electrodes, etc... The dream configuration would allow micronuclei assays to be done first, and comets later on the same embedded cells. In the context of the OpenFlexure scope, we may simply modify the upper stage attachment, to become the 'cheek cell chip' in fact. But first we need to see more than just nuclei with this microscope/RaspberryPi system (work in progress!)!
More to come soon!!
Back to the Genomic integrity project page at the Hackuarium
To the AGiR! site